Maladie de Charcot : 150 ans après sa découverte, que savons-nous ?
Publié par Inserm iBraiN Université de Tours, le 3 octobre 2018 2.7k

Patrick Vourc'h, Université de Tours; Christian Andres, Université de Tours; Philippe Corcia, Université de Tours et Philippe Couratier, Université de Limoges

Cet article est publié dans le cadre de la Fête de la Science 2018 dont The Conversation France est partenaire. Retrouvez tous les débats et les événements de votre région sur le site Fetedelascience.fr
Quel est le lien entre le physicien théoricien et cosmologiste Stephen Hawking, l’écrivain et scénariste Sam Shepard, et l’acteur et gentleman britannique David Niven ? Tous ont fait preuve de génie ou de talent, et ont été récompensés par de prestigieux prix scientifiques, un Prix Pulitzer, ou un Academy Award. Tous ont également souffert d’une même maladie, la sclérose latérale amyotrophique (SLA).
La plus courante des maladies rares
Une maladie est dite « rare » si elle affecte moins d’une personne sur 2 500, ce qui équivaut en France à moins de 30 000 personnes atteintes. C’est ce que l’on appelle la prévalence de la maladie. Aujourd’hui la SLA affecte en France 8 000 personnes. La SLA est donc bien une maladie rare, mais il s’agit toutefois de la plus fréquente des maladies rares. Plus de 1 000 nouveaux cas de SLA sont diagnostiqués chaque année dans notre pays, ce qui représente 4 nouveaux cas par jour (ces chiffres correspondent à l’incidence de la maladie).
Cette maladie est aussi appelée Maladie de Charcot, du nom du neurologue et professeur d’anatomie pathologique Jean Martin Charcot, qui l’a décrite dans les années 1860 en France. Aux États-Unis elle est aussi connue sous l’appellation de maladie de Lou Gehrig (Lou Gehrig’s disease), du nom du fameux joueur de base-ball des Yankees décédé de cette maladie en 1939.
La SLA est une maladie neurodégénérative se caractérisant par la mort des neurones impliqués dans la motricité : les neurones moteurs (ou motoneurones), qui relient le cerveau et la moelle épinière aux muscles du corps. Ces cellules nerveuses sont localisées soit dans le cerveau (motoneurones centraux), soit dans le tronc cérébral et la moelle épinière (motoneurones périphériques). Leur mort se traduit par un durcissement (Sclérose) de la partie latérale (latérale) de la moelle épinière et une fonte des muscles (Amyotrophique).
Il ne faut pas confondre la SLA avec une autre maladie dont le nom est en partie similaire, la sclérose en plaques (SEP).
Sclérose latérale amyotrophique et sclérose en plaques : des symptômes et des causes différentes
Si la sclérose latérale amyotrophique et la sclérose en plaques sont toutes deux des maladies neurodégénératives, la comparaison s’arrête là : leurs symptômes et leurs causes sont en effet différents.
Les premiers symptômes d’une SLA sont souvent des crampes et des secousses musculaires (fasciculations), ou parfois des difficultés à avaler ou à parler. La maladie évolue ensuite vers des difficultés d’usage des bras et des jambes, puis une paralysie. Chez 80 % des patients, la dégénérescence est rapide, et la maladie est responsable d’un décès dans les 3 à 5 ans après la manifestation des premiers symptômes.
La SEP, quant à elle, associe divers symptômes, comme des sensations de picotements, d’engourdissements, de faiblesse musculaire, des pertes d’équilibre, lesquels sont souvent accompagnés de fatigue. Contrairement à la SLA, la SEP n’est pas fatale. L’espérance de vie d’une personne atteinte de SEP est en effet proche de celle de la population générale.
Les causes de la dégénérescence des neurones dans la SLA et la SEP sont également différentes. La SLA résulte de la mort des neurones moteurs, aussi appelés motoneurones, dans le cerveau et la moelle épinière. Les causes de cette mort sont encore mal connues, elles apparaissent multiples. Les motoneurones présenteraient en effet des problèmes de production d’énergie, de transport de substances à l’intérieur de leurs longs prolongements (axones), et de communication (synapses) avec les neurones voisins et les muscles.
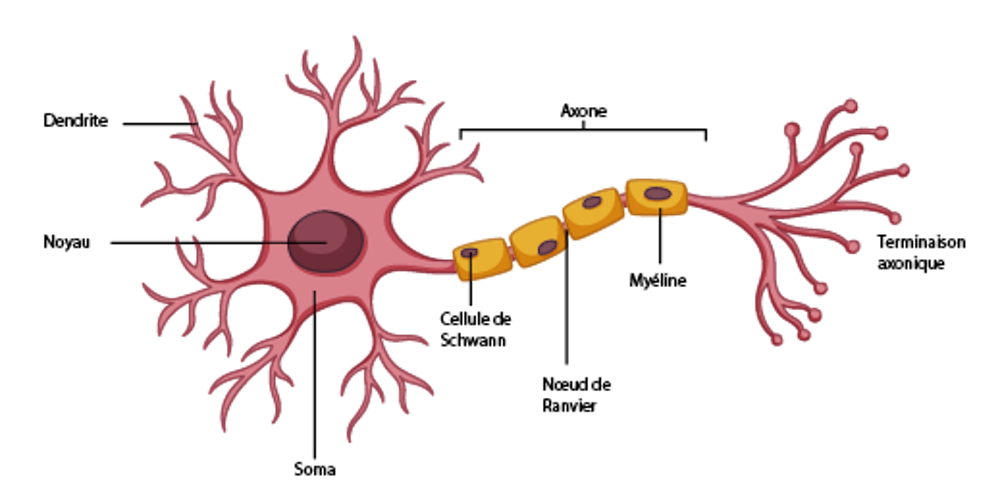
La SEP se définit quant à elle par l’apparition de lésions, aussi appelées plaques, qui apparaissent dans le cerveau, la moelle ou les nerfs optiques, et affectent donc diverses catégories de neurones. Il s’agit d’une maladie auto-immune : le système immunitaire dysfonctionne et s’attaque aux composants normaux de l’organisme, en l’occurrence la couche graisseuse qui entoure les axones, ce qui entraîne la mort des neurones. Cette couche, appelée gaine de myéline, joue un rôle d’isolant qui permet d’accélérer le transport du signal électrique le long des axones.
Une seule maladie, mais des causes multiples
La SLA est une maladie dite complexe. Elle résulte en effet de diverses causes, qui s’ajoutent les unes aux autres jusqu’à atteindre un seuil déclencheur de la maladie. Les scientifiques parlent alors d’étiologie multifactorielle, et nomment « facteurs de risques » ces causes qui s’additionnent chez un individu donné. Les facteurs de risques de la SLA sont :
un âge avancé : la SLA apparaît souvent entre 55 et 60 ans, un âge associé à une certaine « fatigue » des neurones caractérisée par une communication moins efficace entre neurones, du fait de connexions altérées ;
des facteurs environnementaux : ceux-ci sont encore mal identifiés. On peut toutefois citer l’exposition à des métaux lourds et à certaines toxines, mais cela doit être confirmé ;
des facteurs génétiques propres à chaque individu : ces facteurs ont vu leur nombre croître de manière exponentielle depuis 1993, date de la découverte du premier gène impliqué dans la maladie.
La SLA n’est généralement pas familiale
Dans 9 cas sur 10, la SLA apparaît isolée dans une famille, chez une seule personne. On parle alors de SLA sporadique. Les causes de ces SLA sporadiques combinent l’âge, des facteurs environnementaux, métaboliques et des facteurs génétiques.
Dans 1 cas sur 10, la maladie correspond à une forme familiale : on considère qu’une SLA est familiale lorsqu’au moins deux personnes au sein d’une même famille ont une SLA (ou l’ont eu et en sont décédées). Ces SLA dites familiales sont causées par une anomalie d’un ou de quelques gènes, parmi la vingtaine de gènes connus pour être impliqués dans la maladie. Dans la moitié des familles dont des membres ont présenté une SLA, une cause génétique est identifiée.
La lutte se poursuit
La mobilisation de nombreux acteurs autour des chercheurs, comme les associations impliquant les patients (telles que l’ARSLA), a permis de mieux cerner la maladie et ses causes. Ces nouvelles connaissances aboutissent à la mise en place de nombreux programmes de recherche, comme le programme Intrabals de l’Unité 1253 de l’Inserm, Université de Tours. Financé par la Région Centre Val-de-Loire en lien avec le Laboratoire d’Excellence (Labex) MabImprove, il a pour objectif de bloquer l’accumulation toxique d’une protéine dans les motoneurones grâce à des anticorps particuliers, appartenant à la catégorie des intracorps (anticorps produits et demeurant à l’intérieur des cellules).
Par ailleurs, des études internationales sont en cours pour identifier de nouveaux facteurs génétiques de la maladie, telle que l’étude MINE à laquelle participe l’Unité 1253 et les Centres Maladies Rares sur la SLA du CHU de Tours et de Limoges (Filière de soins Filslan.
Plus de 150 ans après sa description par Jean Martin Charcot, la SLA demeure incurable. Néanmoins, grâce à cette mobilisation, de nouvelles pistes de recherche sont explorées en vue d’aboutir le plus rapidement possible à des traitements efficaces.![]()
Patrick Vourc'h, Professeur des Universités - Praticien Hospitalier (PU-PH), HDR, Université de Tours; Christian Andres, Enseignant chercheur, Université de Tours; Philippe Corcia, Enseignant chercheur, Université de Tours et Philippe Couratier, Coordinateur de centre expert, Université de Limoges
La version originale de cet article a été publiée sur The Conversation.
>> Image de Une : L'éminent astrophysicien Stephen Hawking, décédé en avril 2018, a souffert d'une forme rare de SLA, de début précoce et d'évolution lente. Andrew Cowie / AFP.